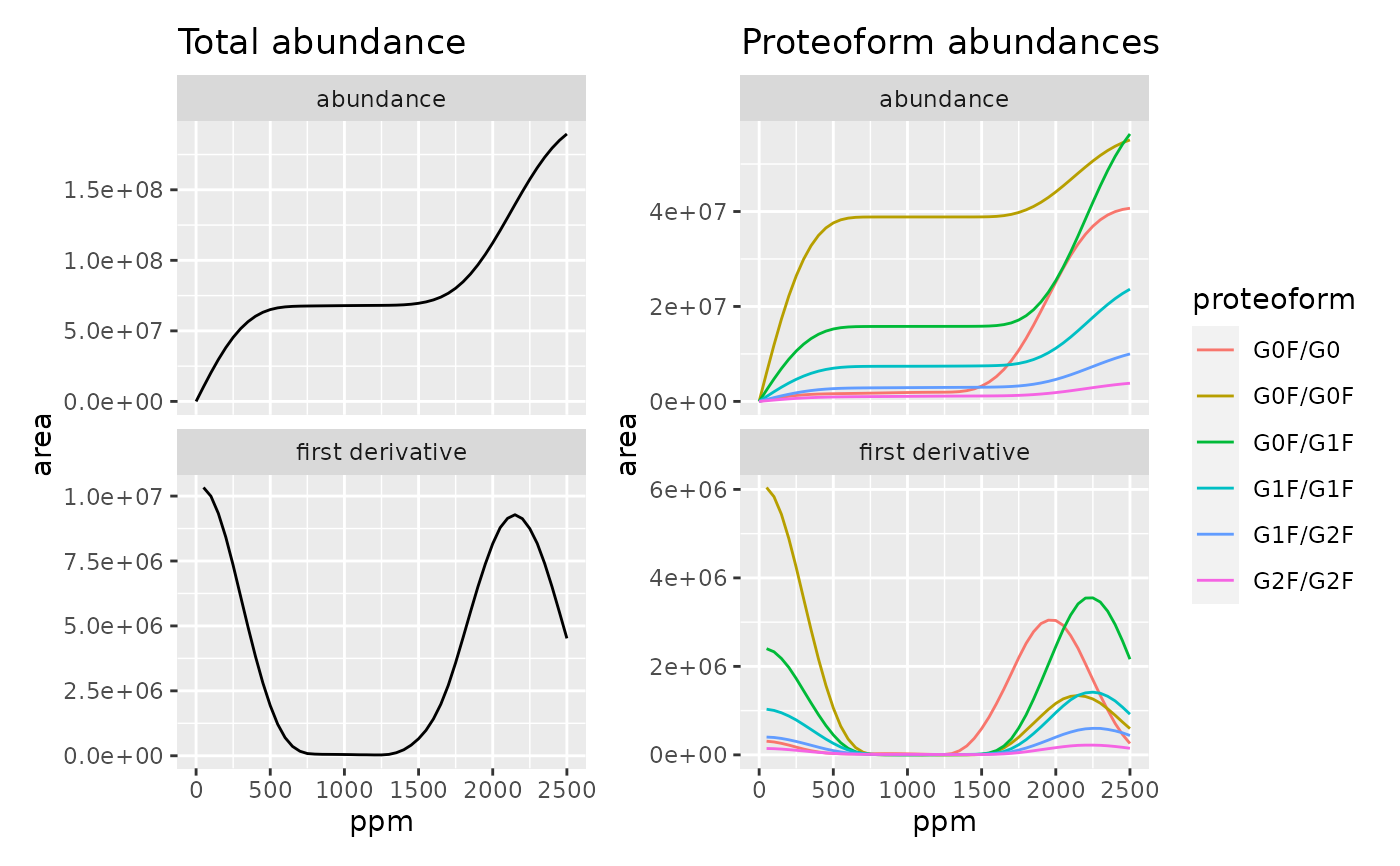
Diagnostic plot for selecting optimal peak integration boundaries.
Source:R/visualization.R
plot_optimal_ppm.RdThis function draws proteoform abundances vs the PPM value used by
ionize(), which in turn determines the integration boundaries for each ion.
plot_optimal_ppm( ms_data, proteoforms, charge_states, ppm_values = seq(0, 2500, 50), rt_limits = c(-Inf, +Inf), subplots = c("all", "total", "modcom") )
Arguments
| ms_data | Mass spectrometry data stored in an mzR object as returned by
|
|---|---|
| proteoforms | Proteoform masses as returned by |
| charge_states | Vector of charge states in which the proteoforms should be quantified. |
| ppm_values | Vectors of PPM values where abundances should be sampled. |
| rt_limits | A specification of retention time limits, passed to
|
| subplots | Subplots that should be drawn; one of |
Value
A ggplot object describing the created plot.
Details
By default, the function creates four plots: Total proteoform abundance vs PPM (upper left), first derivative of this curve (lower left), individual proteoform abundances vs PPM (upper right) and first derivative of the latter curve (lower right).
Optimal integration boundaries capture each peak completely and do not lead to overlaps with neighboring peaks. This corresponds to the PPM value where the curves in the upper plots reach the first plateau, and their derivatives become zero.
Examples
ms_data <- mzR::openMSfile( system.file("extdata", "mzml", "mab1.mzML", package = "fragquaxi") ) proteins <- define_proteins( system.file("extdata", "mab_sequence.fasta", package = "fragquaxi"), .disulfides = 16 ) modcoms <- define_ptm_compositions(sample_modcoms) pfm_masses <- assemble_proteoforms(proteins, modcoms) plot_optimal_ppm(ms_data, pfm_masses, charge_states = 33L:40L, rt_limits = c(300,350))